
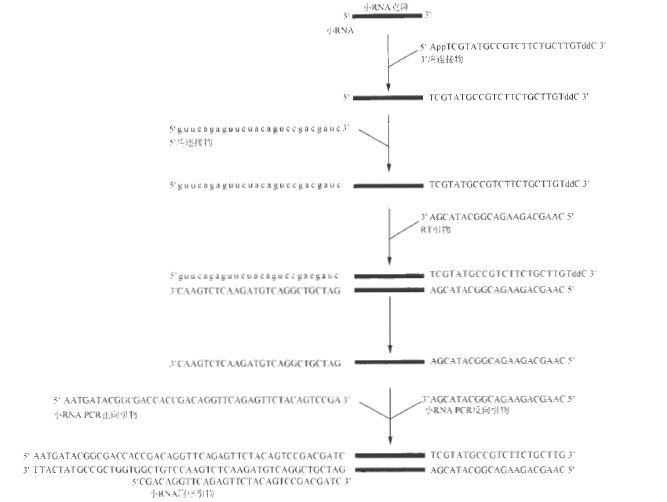
小RNA 克隆和高通量测序的结合是纯化和鉴别细胞或组织中小RNA 的有效先进技术。本方案描述了用于Illumina Genome Analyzer 高通量分析的19 ~ 29 个核苷酸小RNA 的克隆方法,通过聚丙烯酰胺凝胶从总RNA 样品中分离纯化小RNA, 并进行两步连接反应:小RNA3′ 端与5′ 腺苷酸DNA 连接物的连接;以及小RNA5′ 端与RNA 连接物的连接。此后,通过反转录和PCR 制备高通量测序所需的小RNA cDNA 文库。图1 中的流程描述了建立cDNA 文库的具体步骤,图 2 描述了建立小RNA 文库的具体步骤。

试剂
丙烯酰胺: 双丙烯酰胺( 19 : 1, 40%, m/V)
过硫酸铁( 10%, m/V)
绷砂/硐酸缓冲液( 5X, pH8.6 )
氯仿
二甲基亚砜( DMSO )
DNA 分子质量标准(100bp )
dNTP 混合物(每种浓度为10mmol/L)
3’端连接反应缓冲液
乙醇(100%和80% )
溴化乙锭
Ficoll-Orange G 上样缓冲液( 6 X)
甲酰胺上样缓冲液
肝糖原( 20μg/μL)
连接物: 3’ 端连接物( 5′-rAppTCGTATGCCGTCTTCTGCTTGT/ddc/-3′)
5’端连接物( 5′-GUUCAGAGUUCUACAGUCCGACGAUC-3′)
M13 的正、反向引物
mirVana miRNA 分离提取试剂盒或TRizol 试剂
GTG 琼脂糖凝胶
苯酚(pH7.9)
酚: 氯仿: 异戊醇( 25 : 24 : 1, V/ V/ V, pH 8.0 )
PuReTaq Ready-To-Go PCR Beads
反转录引物( 5′-CAAGCAGAAGACGGCATACGA-3′)
RNA Century Marker
总RNA 样品
小RNA PCR 正向引物( 5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACA
GTCCGA-3′)
小RNAPCR 反向引物( 5′-CAAGCAGAAGACGGCATACGA-3′ )
乙酸钠(3mol/L, pH5.2 )
氯化钠( 0.3mol/L 和0.6mol/L)
高碘酸钠( 200mmol/ L)
Superscript III 反转录酶
SYER Gold ( 10 000 X)
合成的小RNA 分子质量标准( 18 个、30 个和60 个核苷酸)
T4RNA 连接酶2 ,截短的或T4RNA 连接酶2 , 截短的K2276
T4RNA 连接酶和10XT4RNA 连接酶缓冲液
TAE 缓冲液( 50 X 和1 X)
TBE 缓冲液(5X 和0.5X)
四甲基乙二胺( TEMED )
TOPOTA 克隆试剂盒
尿素
设备
离心机
玻璃托盘
加热模块
微量离心管(2.0mL, 1.5mL)
NanoDrop 分光光度计
PCR 管( 0.5mL)
聚丙烯酰胺凝胶装置
刀片(新)
Thermo 热循环仪
紫外灯(302nm)
涡旋振荡仪
方法
总RNA 纯化
1 按照使用说明书,用mirVana miRNA 分离提取试剂盒或TRlzol 试剂纯化总RNA, 并溶解于水中。
2 通过紫外分光光度计检测RNA 的浓度和纯度。
从总RNA 中纯化l9 ~ 29 个核苷酸的小RNA
3 配制含8mol/L尿嘧啶的15 %变性聚丙烯酰胺凝胶(长20cm X 宽16cm X 厚1.5mm ),用6 孔梳子(左右两边的两个小孔,深17mmX 宽5mmX 厚1.5mm, 小孔上样分子质量标准,中间4 个大孔用来上样品,深17mmX 宽30mmX 厚1.5mm)
4 在0.5 X TBE 缓冲液中35W 恒定功率预电泳30 min 。
5 用50μL 水稀释100μg 总RNA, 并加入等体积的甲酰胺上样缓冲液进行混合。用甲酰胺上样缓冲液稀释人工合成的18 核苷酸和30 核苷酸RNA 寡核苷酸,并配制成终浓度为1μmol/L, 用作小RNA 分子质量标准。
6 95 °C 加热样品和分子质量标准5min, 然后置冰上冷却。
7 洗涤聚丙烯酰胺凝胶孔以洗去尿嘧啶,每个大孔中加入总RNA 样品100μL(步骤5) 。聚丙烯酰胺凝胶边缘小孔加入10μL (10pmol 每孔)小RNA 分子质量标准混合液(来自步骤5) 。
8 0.5 X TBE 缓冲液中恒定功率电泳,至溴酚蓝距离孔缘约6cm 。
9 电泳完成后,室温下用SYBR Gold (用0.5 X TBE 缓冲液1 : 10 000 稀释)将胶固定在洁净的玻璃托盘中5min 。将胶固定于玻璃托盘之前必须在托盘中加入染色剂。
10 在302nm 紫外灯下观察RNA , 用新刀片切下18 ~ 30 个核苷酸之间且包含RNA 的凝胶,并将切下的凝胶放入预先称量好的2 .0mL 微量离心管中。
11 每100mg 凝胶中加入300μL 0.3mol/L 氯化钠,室温下旋转过夜以洗脱小RNA 。
12 加入1μL 20μg/μL 糖原和3 倍体积的无水乙醇沉淀小RNA 。涡旋混合,将混合液置于-20 °C 或-20 °C 以下保存3h 。4 °C 最大转速离心30min, 收集底部沉淀。
13 用1mL80%乙醇洗涤沉淀1 次,洗去残留盐分。4 °C 最大转速离心15min 以回收小RNA 。打开离心管管口,在实验台上静置数分钟,尽可能除去80%乙醇。
14 将沉淀物溶解于13μL 无核酸酶的水中。
氧化法富集3′ 端修饰的小RNA (可选)
与动物miRNA 不同,动物Piwi 相互作用RNA( piRNA) 和苍蝇内源性siRNA 的3’端都被2′-O-甲基化,从而降低其与3’端连接物的反应效率。此外,相同细胞中2′-O- 甲基化的piRNA 或siRNA 丰度通常较miRNA 丰度低。通过氧化反应将miRNA 末端的2′, 3’羟基氧化成醛基,从而阻断其与3’ 端连接物的连接,并增强2′-O- 甲基化小RNA 的测序效率。
15 依次加入下列试剂,建立40μL 的氧化反应体系:

16 移液管轻轻吹打混合溶液,离心1 ~ 2 s 收集试管底部反应物。
17 反应物25 °C 孵育30min 。
18 加入229μL 水、1μL 20μg/μL 糖原以及3mol/L 乙酸钠( pH5.2) 30μL 。移液管轻
轻吹打混匀,并加入3 倍体积的无水乙醇。涡旋以混合溶液,-20 °C 或-20 °C 以下保存过夜。
19 4 °C 最大转速离心30min, 收集试管底部沉淀。
20 用1mL 80%乙醇洗涤沉淀物以清除残留盐分。4 °C 最大转速离心15min 以回收小RNA 。打开管口,实验台上静置数分钟,使80%乙醇尽可能挥发干净。
21 将沉淀物溶解于13μL 无核酸酶的水中。
小RNA 与3’端连接物连接
22 依次加入以下试剂,建立20μl 的3’端连接反应体系:

23 为准确检测3’端连接的小RNA, 需依次加入以下试剂,配制3’端连接分子质量
标准。

24 轻轻敲打试管A 和B 外壁,混匀。离心1 ~ 2s, 使试剂置于试管底部。
25 试管A 和B 均4 °C 过夜。
26 试管A 中加入20μL 甲酰胺上样缓冲液(来自步骤25) 。
27 试管B 中加入180μL 甲酰胺上样缓冲液,配制3 端连接分子质量标准( 来自步骤25) 。
28 试管A 和B 于95 °C 加热5min, 置冰上冷却。
3’端连接小RNA 纯化
29 配制含8mol/L 尿嘧啶的15 %变性聚丙烯酰胺凝胶( 长20cmX 16cmX 1.5mm) ,用20 孔梳子(孔大小: 深17mmX 宽5mmX 厚1.5mm) ( 方案7) 。
30 0.5XTBE 缓冲液中35W 恒定功率预电脉30min 。
31 洗去胶孔中的尿素,每孔加入40μL 3’端反应样品(来自步骤28) 。不同样品之间至少空一个孔,避免交叉污染。胶左右两侧边缘孔上样40μL3 端连接分子质量标准(来自步骤28) 。
32 0.5 X TBE 缓冲液恒定功率20W 电泳,至溴酚蓝移至胶底部。
33 按照步骤9 所描述的方法将胶固定好。
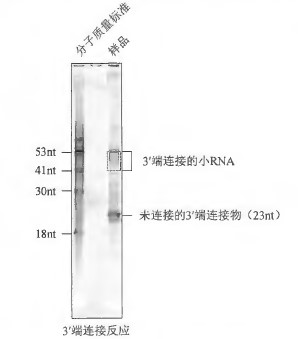
34 302nm 紫外灯下观察RNA, 对应在两个分子质量标准之间的位置,用新刀片切下包含RNA 的凝胶(图3)。将切下的凝胶放入预先称重的1.5mL 微量离心管中。
35 如步骤11 ~步骤14 所述,对3′ 端连接的小RNA 进行洗脱、沉淀并溶解。
连接小RNA与5’端连接物
36 在1.5mL 的微量离心管中,一次加入下列试剂,建立20μL 5′ 端连接反应体系:

37 轻轻敲打试管外壁,混匀。离心1 ~ 2s, 使试剂处于试管底部。
38 4℃过夜。
39 加入等体积的甲酰胺上样缓冲液, 终止反应。95°C 加热5min, 置冰上冷却。
5’端连接小RNA 纯化
40 如步骤29 和步骤30 所描述,配制10%的变性聚丙烯酰胺凝胶并预电泳。
41 向78μL 甲酰胺上样缓冲液中加入1μL ( 10pmol) 合成的60 个核酸的RNA 寡核苷
酸与1μL (1μg) RNA Century 分子质量标准,配制成RNA 分子质量标准。95°C 加热5min,置冰上冷却。
42 洗去胶孔中的尿素,向各个孔中加入40μL 5′ 端连接反应样品(步骤39 ) ,不同样品之间至少空一个孔,避免交叉污染。左右两侧边缘孔中加入40μLRNA 分子质量标准。
43 如步骤32 和步骤33 所述,电泳并固定。
44 302nm 紫外灯下观察RNA, 并用新刀片切下60 ~ 100 核苷酸包含RNA 的凝胶,
放入预先沉重的1.5mL 微量离心管中。
45 如步骤11 ~ 步骤13 所述,洗脱并沉淀5′ 端连接的小RNA 。
46 将沉淀物溶于20μL 水中。
反转录
47 向含有20μL 5′ 端连接的小RNA (来自步骤46) 试管中加入1 μL (100pmol) 反转
录引物。
48 72 °C 孵育2min 。
49 室温下最大转速离心lmin, 置冰上冷却。
50 混合以下试剂,配制RT 混合液:

51 向试管中加入16.8μL RT 混合液,并将其分装成两管:+ RT 和- RT, 每管18μL 。
52 向+ RT 管中加入2μL (400U) 的SuperScript 反转录酶Ⅲ ,向- RT 管中加入2μL 水。
53 50 °C 孵育1h,再70 °C 加热15min 灭活反应。
cDNA 文库扩增
54 混合以下试剂,配制引物混合物:

55 在3 个含有PuReTaq Ready-To-Go PCR Bead 的0.5mLPCR 试管中,每管分别加入23μL 的引物混合物。
56 向3 个PCR 管中分别加入2μL + RT 反应液、-RT 反应液(作为阴性对照,检测
PCR 扩增来自模板而不是其他cDNA) ,或水(作为PCR 阴性对照, 检测PCR 试剂是否污染) 。
57 轻敲试管外壁,混匀溶液。离心1~2s, 使试剂处于试管底部。
58 将试管放入热循环仪中,按下列程序进行cDNA 文库扩增:
i. 94°C 2min
ii. 94 °C 15s
iii. 58 °C 30s
IV. 72 °C 30s
v 重复步骤58ii 和iii 4 个循环或更多。
vi. 94°C 15s
vii. 60 °C 30s
viii. 72 °C 30s
ix 重复步骤vi~v iii 16 个循环或更多。
X. 4 °C 保存
xi. 结束
59 PCR 结束后,向各个PCR 管中加入5μL 6 X Fi coll-Orange G 上样缓冲液,涡旋振荡混合。
60 用含0.5μg/mL 溴化乙锭的1 X TAE 缓冲液配制2% NuSieve GTG 琼脂糖凝胶(长1cm X 宽15cm X 厚12mm ) 。
61 每孔中加入步骤59 中的PCR 样品30μL, 不同样品之间至少空一个孔。
62 左右两侧边缘孔中各加入10μL (1μg) 的100bp DNA 分子质量标准。
63 在1XTAE 缓冲液中洹压100V 电泳,至Orange G 染料移至约胶长3/4 处(约8 0min ) 。
64 302nm 紫外灯下观察条带,用新刀片切下所需DNA 条带并放入预先称重的2.0mL微量离心管中。
65 每100mg 凝胶中加入100μL 0.6mol/L 氯化钠。
66 70 ℃ 孵育至胶完全溶解(约10min) 。
67 加入等体积苯酚(pH 7 . 9 ,预热至室温) 。涡旋振荡20s , 室温下最大转速离心15min,将水层与有机层分离。
68 将水层转移至一个新的1.5mL 微量离心管中,加入等体积酚:氯仿: 异戊醇混合物(25 : 24 : 1, V/V/V, pH 8.0) ,涡旋振荡20s 。室温下最大转速离心15min, 分离水层和有机层。
69 将水层转移至一个新1. 5mL 微量离心管中,加入等体积氯仿,涡旋振荡20s 。室温下最大转速离心15min, 分离水层和有机层。
70 将水层转移至一个新1.5mL 微量离心管中,加入3 倍体积乙醇。涡旋振荡,混匀,-20 °C 或-20 °C 以下放置3h 。4°C 最大转速离心30min, 收集底部沉淀。
71 用lmL80%乙醇洗涤沉淀,除去残余盐分。4 °C 最大转速离心15min, 回收DNA 。打开离心管口并在实验台上静置数分钟,使乙醇尽可能挥发。
72 将DNA 沉淀溶于20μL 无核酸酶的水中,应用NanoDrop 分光光度计检测DNA 浓度。标准浓度范围为20 ~ 50ng/ μL , -2 0 °C 储存DNA 溶液(如小RNA 文库)至下次高通量测序时使用。
Sanger 测序法验证小RNA 文库(可选)
73 按照使用说明书,进行TOPO TA 克隆,每个小RNA 文库中挑取20 个或更多个克隆。
74 应用插入片段两侧的引物( 如Ml3 正向、反向引物),进行菌落PCR 。
75 利用1.5% 琼脂糖凝胶对PCR 产物进行鉴定,然后对PCR 产物逐个克隆进行测序。
疑难解答
问题: 核糖体RNA 交叉污染。
解决方案: 核糖体RNA 交叉污染表明制备的总RNA 可能发生降解。而降解可能主要发生于步骤14 之前。如果起始总RNA 质量没有问题,那么降解可能主要发生于凝胶纯化过程(步骤3 ~步骤11) 。实验全程必须戴手套并经常更换,所用的试剂、试管和移液器必须无RNA 酶污染。RNA 凝胶电泳的TBE 缓冲液和配胶设备必须是独立专用,同时凝胶纯化前必须彻底清洗配胶设备。如果起始总RNA 发生降解,需重新制备高质量的总RNA 。
问题:小RNAcDNA 文库被其他生物小RNA 污染。
解决方案: 这种污染可能主要发生生于凝胶纯化过程中。实验全程必须戴手套并经常更换,凝胶设备和SYBRGold 染色所用托盘在使用之前必须彻底清洗。
问题: Sanger 测序发现,随意挑取的2 0 个克隆中有2 个或2 个以上没有插入小RNA 。
解决方案: 这主要是由于纯化过程中扩增文库被PCR 引物二聚体污染。切胶必须小心,如果有必要,可以用6%~ 10%的非变性聚丙烯酰胺凝胶( 29 : 1, 丙烯酰胺:双丙烯酰胺)代替2% NuSieve GTG 琼脂糖凝胶以提高凝胶分离效果。
配方

